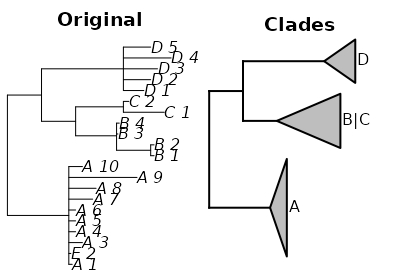
I think the script is finally doing what I wanted.
From the answer that @CactusWoman provided, I changed the code a little bit so the script will try to find the MRCA that represents the largest branch that matches to my search pattern. This solved the problem of not merging non-polytomic branches, or collapsing the whole tree because one matching node was mistakenly outside the correct branch.
In addition, I included a parameter that represents the limit for the pattern abundance ratio in a given branch, so we can select and collapse/group branches that have at least 90% of its tips matching to the search pattern, for example.
library(geiger)
library(phylobase)
library(ape)
#functions
find_best_mrca <- function(phy, group, threshold){
group_matches <- phy$tip.label[grepl(group, phy$tip.label, ignore.case=TRUE)]
group_mrca <- getMRCA(phy,phy$tip.label[grepl(group, phy$tip.label, ignore.case=TRUE)])
group_leaves <- tips(phy, group_mrca)
match_ratio <- length(group_matches)/length(group_leaves)
if( match_ratio < threshold){
#start searching for children nodes that have more than 95% of descendants matching to the search pattern
mrca_children <- descendants(as(phy,"phylo4"), group_mrca, type="all")
i <- 1
new_ratios <- NULL
nleaves <- NULL
names(mrca_children) <- NULL
for(new_mrca in mrca_children){
child_leaves <- tips(tree.test, new_mrca)
child_matches <- grep(group, child_leaves, ignore.case=TRUE)
new_ratios[i] <- length(child_matches)/length(child_leaves)
nleaves[i] <- length(tips(phy, new_mrca))
i <- i+1
}
match_result <- data.frame(mrca_children, new_ratios, nleaves)
match_result_sorted <- match_result[order(-match_result$nleaves,match_result$new_ratios),]
found <- numeric(0);
print(match_result_sorted)
for(line in 1:nrow(match_result_sorted)){
if(match_result_sorted$ new_ratios[line]>=threshold){
return(match_result_sorted$mrca_children[line])
found <- 1
}
}
if(found==0){return(found)}
}else{return(group_mrca)}
}
add_triangle <- function(phy, group,phylo_plot){
group_node_labels <- phy$tip.label[grepl(group, phy$tip.label)]
group_mrca <- getMRCA(phy,group_node_labels)
group_nodes <- descendants(as(tree.test,"phylo4"), group_mrca, type="tips")
names(group_nodes) <- NULL
x<-phylo_plot$xx
y<-phylo_plot$yy
x1 <- max(x[group_nodes])
x2 <-max(x[group_nodes])
x3 <- x[group_mrca]
y1 <- min(y[group_nodes])
y2 <- max(y[group_nodes])
y3 <- y[group_mrca]
xcoords <- c(x1,x2,x3)
ycoords <- c(y1,y2,y3)
polygon(xcoords, ycoords)
return(c(x2,y3))
}
#main
cat("((A_1:0.05,E_2:0.03,A_3:0.2,A_4:0.1,A_5:0.1,A_6:0.1,A_7:0.35,A_8:0.4,A_9:01,A_10:0.2):0.9,((((B_1:0.05,B_2:0.05):0.5,B_3:0.02,B_4:0.04):0.6,(C_1:0.6,C_2:0.08):0.7):0.5,(D_1:0.3,D_2:0.4,D_3:0.5,D_4:0.7,D_5:0.4):1.2):0.5);", file = "ex.tre", sep = "\n")
tree.test <- read.tree("ex.tre")
# Step 1: Find the best MRCA that matches to the keywords or search patten
groups <- c("A", "B|C", "D")
group_labels <- groups
group_edges <- numeric(0)
edge.width <- rep(1, nrow(tree.test$edge))
count <- 1
for(group in groups){
best_mrca <- find_best_mrca(tree.test, group, 0.90)
group_leaves <- tips(tree.test, best_mrca)
groups[count] <- paste(group_leaves, collapse="|")
group_edges <- c(group_edges,best_mrca)
#Step2: Remove the edges of the branches that will be collapsed, so they become invisible
edge.width[tree.test$edge[,1] %in% c(group_edges[count],descendants(as(tree.test,"phylo4"), group_edges[count], type="all")) ] <- 0
count = count +1
}
#Step 3: plot the tree hiding the branches that will be collapsed/grouped
last_plot.phylo <- plot(tree.test, show.tip.label = F, edge.width = edge.width)
#And save a copy of the plot so we can extract the xy coordinates of the nodes
#To get the x & y coordinates of a plotted tree created using plot.phylo
#or plotTree, we can steal from inside tiplabels:
last_phylo_plot<-get("last_plot.phylo",envir=.PlotPhyloEnv)
#Step 4: Add triangles and labels to the collapsed nodes
for(i in 1:length(groups)){
text_coords <- add_triangle(tree.test, groups[i],last_phylo_plot)
text(text_coords[1],text_coords[2],labels=group_labels[i], pos=4)
}
{kind=link}
{kind=link}