Data
I have the following (simplified) dataset, we call df from now on:
species rank value
1 Pseudomonas putida family Pseudomonadaceae
2 Pseudomonas aeruginosa family Pseudomonadaceae
3 Enterobacter xiangfangensis family Enterobacteriaceae
4 Salmonella enterica family Enterobacteriaceae
5 Klebsiella pneumoniae family Enterobacteriaceae
6 Pseudomonas putida genus Pseudomonas
7 Pseudomonas aeruginosa genus Pseudomonas
8 Enterobacter xiangfangensis genus Enterobacter
9 Salmonella enterica genus Salmonella
10 Klebsiella pneumoniae genus Klebsiella
11 Pseudomonas putida species Pseudomonas putida
12 Pseudomonas aeruginosa species Pseudomonas aeruginosa
13 Enterobacter xiangfangensis species Enterobacter hormaechei
14 Salmonella enterica species Salmonella enterica
15 Klebsiella pneumoniae species Klebsiella pneumoniae
What I want to achieve
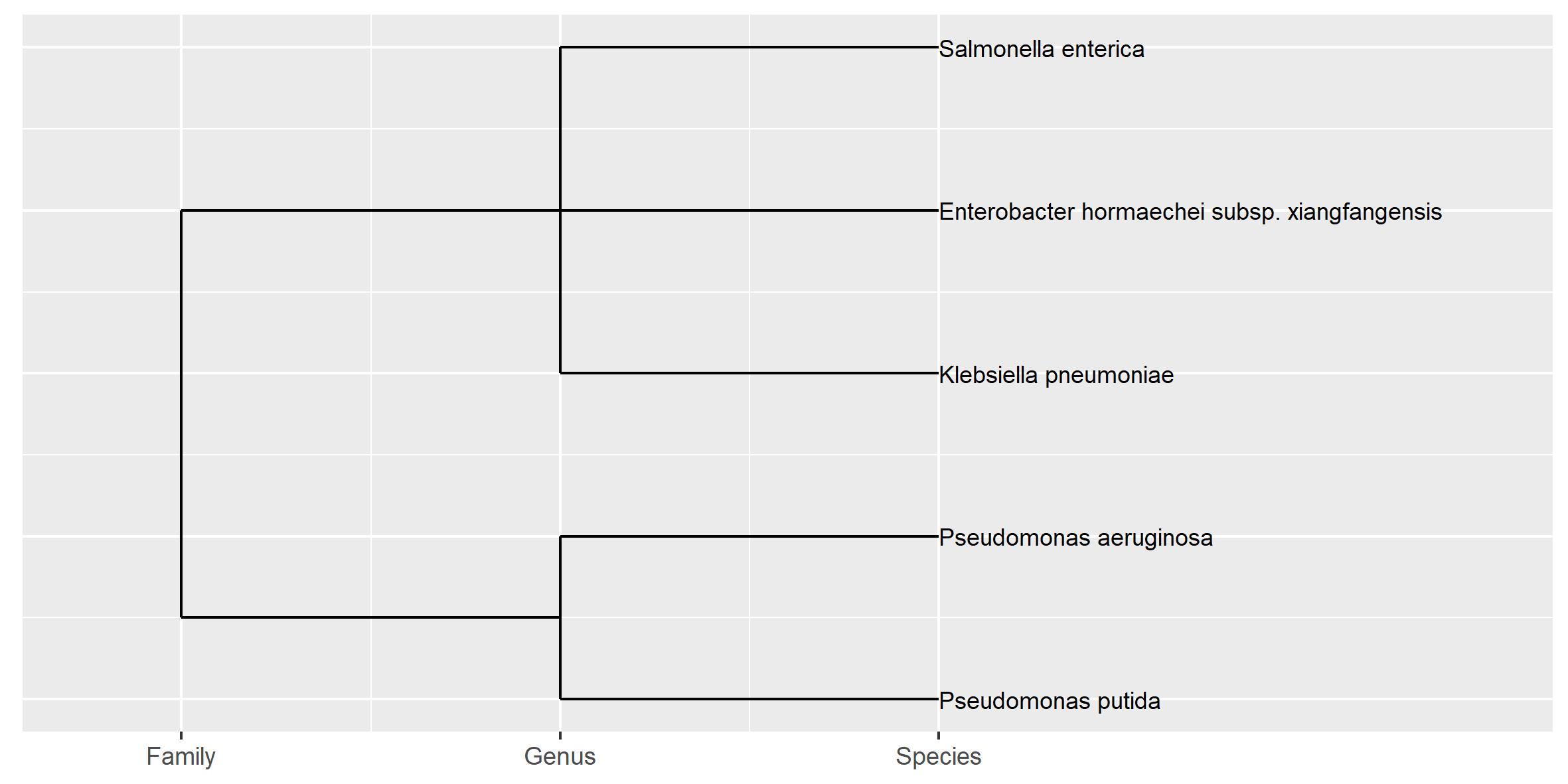
This data is taxonomy data that shows the species classification, where the rank is in order of family > genus > species. Due to the hierarchical nature I want to show this as a tree, preferentially in ggplot2 like so:
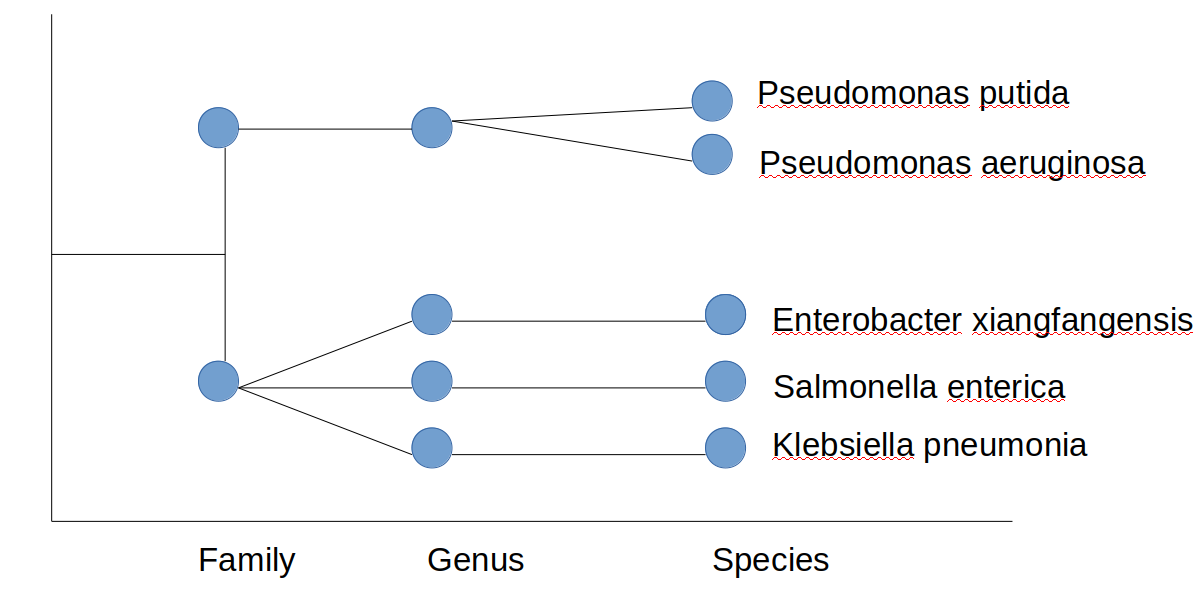
What I tried
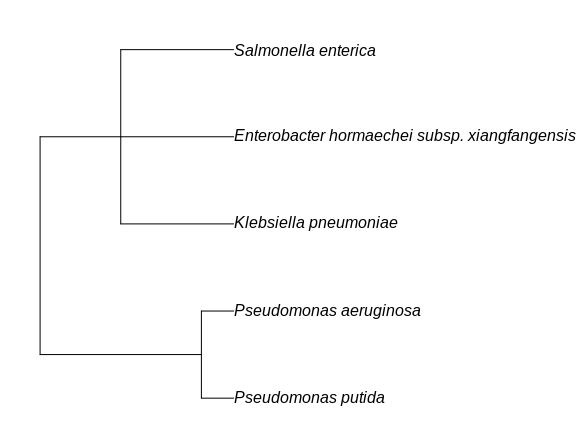
While I found a package, taxize written to convert this (actually the full classification - only partially shown here) to a tree, using class2tree:
class.dat <- classification(c("Pseudomonas putida", "Pseudomonas aeruginosa","Enterobacter xiangfangensis","Salmonella enterica","Klebsiella pneumoniae"), db = 'ncbi')
taxize::class2tree(class.dat)
This does not show the ranks like in my hand made graph, that I need in my visualization:
EDIT: dput of data
structure(list(species = c("Pseudomonas putida", "Pseudomonas putida",
"Pseudomonas putida", "Pseudomonas aeruginosa", "Pseudomonas aeruginosa",
"Pseudomonas aeruginosa", "Enterobacter xiangfangensis", "Enterobacter xiangfangensis",
"Enterobacter xiangfangensis", "Salmonella enterica", "Salmonella enterica",
"Salmonella enterica", "Klebsiella pneumoniae", "Klebsiella pneumoniae",
"Klebsiella pneumoniae"), rank = c("family", "genus", "species",
"family", "genus", "species", "family", "genus", "species", "family",
"genus", "species", "family", "genus", "species"), value = c("Pseudomonadaceae",
"Pseudomonas", "Pseudomonas putida", "Pseudomonadaceae", "Pseudomonas",
"Pseudomonas aeruginosa", "Enterobacteriaceae", "Enterobacter",
"Enterobacter hormaechei", "Enterobacteriaceae", "Salmonella",
"Salmonella enterica", "Enterobacteriaceae", "Klebsiella", "Klebsiella pneumoniae"
)), row.names = c(NA, -15L), class = "data.frame", .Names = c("species",
"rank", "value"))
EDIT: Response to @StupidWolf
I was able to convert the class.data to a dataframe and then into a parent-child dataframe to use it as input for the ggraph. The only thing left is having the xlabel, in this case the interest vector. However I'm not sure if that's possible in ggraph:
# Retreive data
class.dat <- classification(c("Pseudomonas putida", "Pseudomonas aeruginosa","Enterobacter xiangfangensis","Salmonella enterica","Klebsiella pneumoniae"), db = 'ncbi')
# Specify interest
interest <- c('superkingdom', 'phylum','class','order','genus','species')
# Convert to wide matrix
df2 <- bind_rows(class.dat, .id = "column_label") %>%
dplyr::select(-id) %>%
filter(rank %in% interest) %>%
spread(rank, name) %>%
dplyr::select(-column_label) %>%
dplyr::select(interest) %>% # we need the order
as.matrix()
# Empty parent child matrix
parent.child <- matrix(nrow=0,ncol=2)
# Add data to parent child
for (i in 1:(ncol(df2)-1)){
parent.child <- rbind(parent.child,df2[,c(i,i+1)])
}
# To dataframe and add colnmaes
parent.child <- as.data.frame(parent.child)
colnames(parent.child) <- c('from', 'to')
# Convert this to a ggraph
g <- graph_from_data_frame(parent.child)
ggraph(g,layout='dendrogram',circular=FALSE) +
geom_edge_link() +
geom_node_label(aes(label=names(V(g))),size=3,nudge_y=-0.1) +
scale_y_reverse(labels = interest) + coord_flip() +
theme_classic()
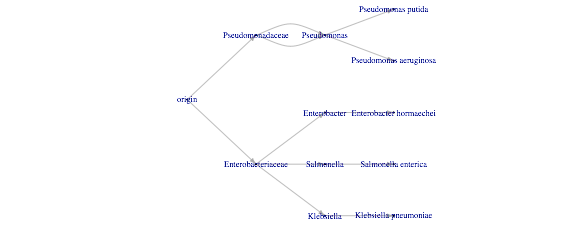

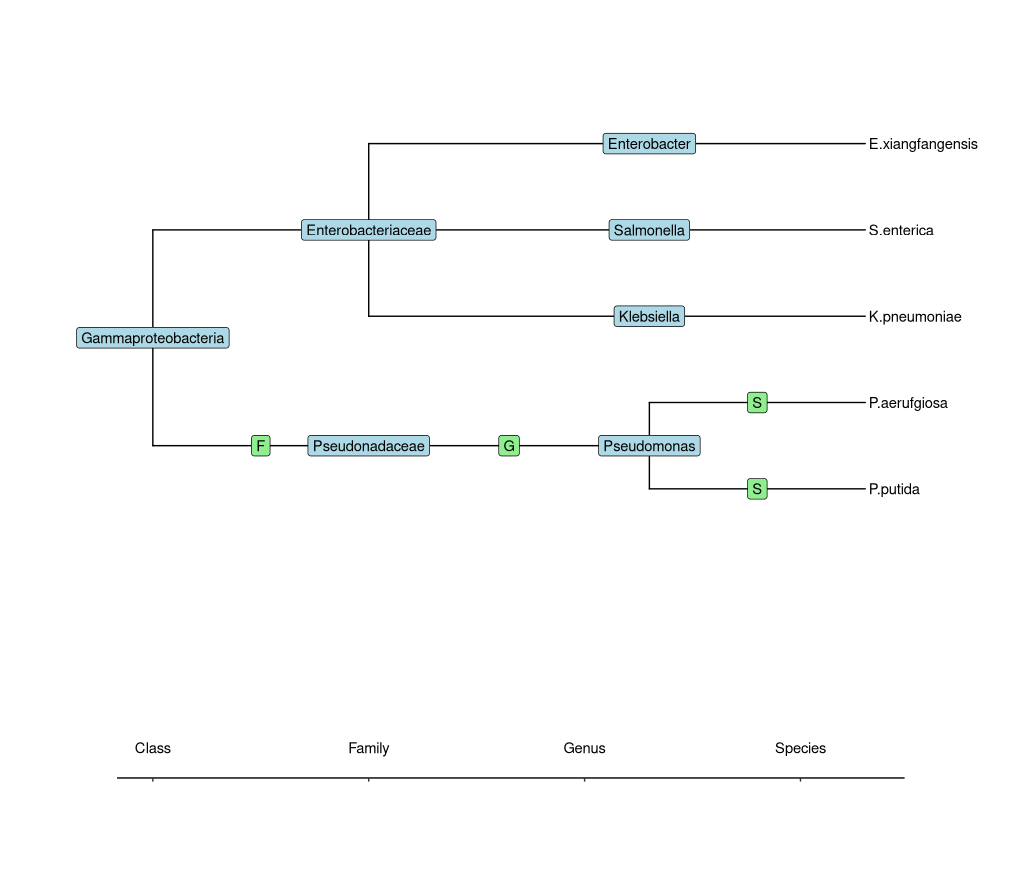
dputof your data? – Primateshiptaxizeand thereby not explicitly show at whatranka species deviates – Proportioned